Intro to Nextflow: users
You know those times when you’re working on a massive research project, and datasets just start comin’ and they don’t stop comin’? Yeah, those times are when a tool like Nextflow can be a serious lifesaver. Nextflow is an open-source pipelining tool that makes processing these kinds of massive computational workflows somewhat easy.
One of the best things about Nextflow is that it takes care of all the annoying little details that can bog you down when you’re working with complex data in different environments. This means you can focus on the actual scientific work you’re doing, instead of worrying about managing the infrastructure behind it all. Plus, Nextflow is great for a variety of scientific fields, like genomics, radiomics, and other high-throughput data analysis.
But of course, you still need to know a bit about what you’re doing. You’ll want to brush up on your scripting knowledge and be comfortable working with things like clusters, containers, and cloud platforms. And of course, you’ll need to learn Nextflow’s syntax and features.
Using Nextflow can make a huge difference in how quickly and efficiently, you’re able to conduct your research. It’s a game-changer, and definitely not something you want to overlook if you’re serious about large data processing tasks. Nextflow knows how much RAM and how much CPU each process needs and which tasks must be done first to allow which one next. So, Nextflow optimize the input/ouput and transfer the information in order to keep your CPU running as close to 100% as possible.
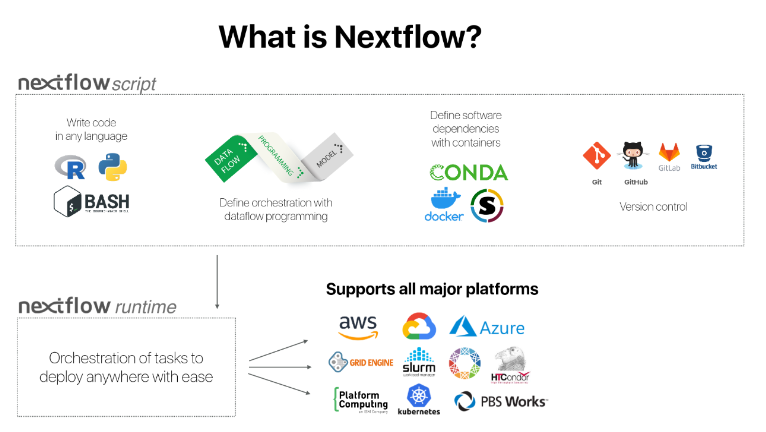
Nextflow is simply a tool that helps users to create efficient pipeline, assembling processes, facilitate logging, organize your files, and launch large processing jobs on a variety of infracstructures.
Installation
First, Nextflow can be used on any Linux and MacOS. It requires Bash 3.2 (or later) and Java 11 (or later, up to 20) to be installed. You can find the right Java version, then install it for your system.
It is common to explicitely tell Nextflow where Java is. Use this command: readlink -f `which javac` | sed "s:/bin/javac::" to get the full path. Then, add this line to your .bashrc: export JAVA_HOME=/PATH/FOR/JAVA/YOU/JUST/GOT
These instructions are for Nextflow v21.12, if needed you can pick another Nextflow release here. Make sure to execute this in a directory you will remember (likely next to the rest of your tools).
wget https://github.com/nextflow-io/nextflow/releases/download/v21.12.1-edge/nextflow-21.12.1-edge-all
mv nextflow-21.12.1-edge-all nextflow
chmod +x nextflow
./nextflow run hello
To execute nextflow from anywhere, add the directory where you downloaded it to your PATH in your .bashrc: export PATH=/PATH/TO/NEXTFLOW:${PATH}
Overview
Usage & Help
Assuming you installed Nextflow (and most other software)., you can get the help to display by typing:
nextflow /PATH/TO/PIPELINE/main.nf --help
The lab has multiple Nextflow pipeline (Tractoflow, Connectoflow, RBx-Flow, Freesurfer-Flow, etc.), and they all have a very similar organization.
First thing to know is how to get the code. All pipeline are available on the Scilus Github organization page. Our most well-known and well-organized pipeline is Tractoflow, so let’s use this one as an example.
- Once cloned, there is 4 files to understand:
README.mdcontains the important information about the repository, authors, documentation, references, etc.USAGEcontains the explicit documentation about how to write the command line, all mandatory and optional parameters and to actually launch the pipelinenextflow.configcontains all the default values for mandatory and optional parametersmain.nfcontains the pipeline itself, all the processes that will be run and how to inter-connect them.
Assuming you installed Nextflow (and most other software) here., you can get the help to display by typing:
nextflow /PATH/TO/PIPELINE/main.nf --help
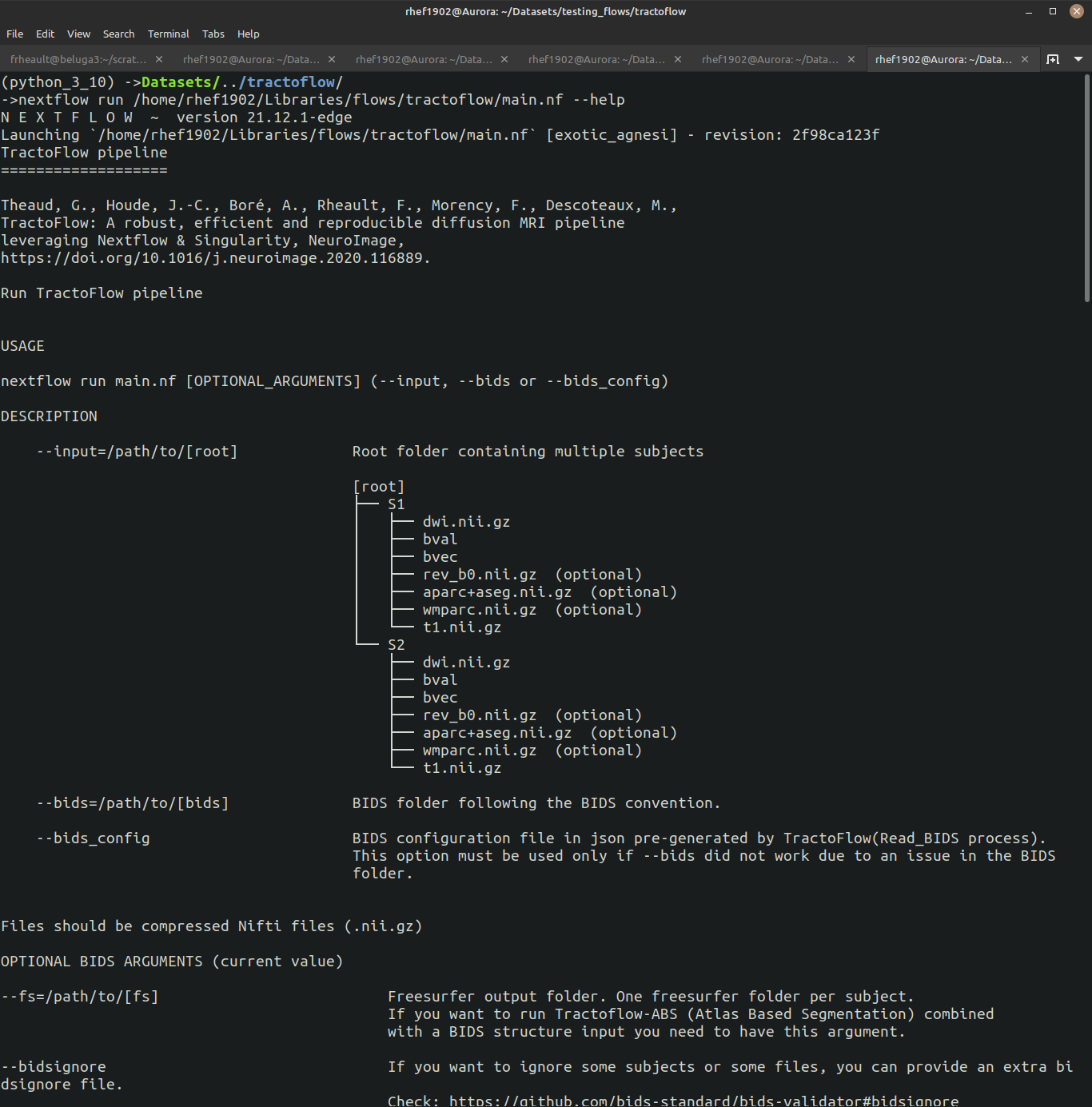
The --help option displays all the relevant information to launch the pipeline correctly. You can often trust the default parameters, but it is crucial to understand what they do and what processes they are impacting.
Tractoflow is a special pipeline, with its own ReadTheDocs and publication we recommend looking at both in detail to understand the tool as well as how to use this pipeline. Running Tractoflow is a great way to get familiar with all tools of the lab, from Nextflow and Singularity to run it, to MI-Brain to visualize outputs and Scilpy to manipulate TRK and NIFTI or bash to move files around after completion.
- Ressources:
Launching it
When it is time to launch, you have to remember that you need to provide all mandatory parameters in the right order.
First, you need to be organized! We suggest cloning all code related to Nextflow in a specific directory (with the rest of your source code), and do your processing in a separated directory. For example, if you are running Tractoflow, considered creating a tractoflow_date_month_year/ directory. Inside of this directory, you can put your data (following Tractoflow specification), a Singularity container, bash script to organize your data, SLURM dispatch file, etc.
All our flows need at least nextflow /PATH/TO/PIPELINE/main.nf --input /PATH/TO/DATA, but then you can (and should) customize any relevant parameters you saw in the USAGE (or –help). All parameters for the pipeline need two dashes (e.g. --input), and all parameters related to Nextflow itself need one dash (e.g. -profile, -with-singularity).
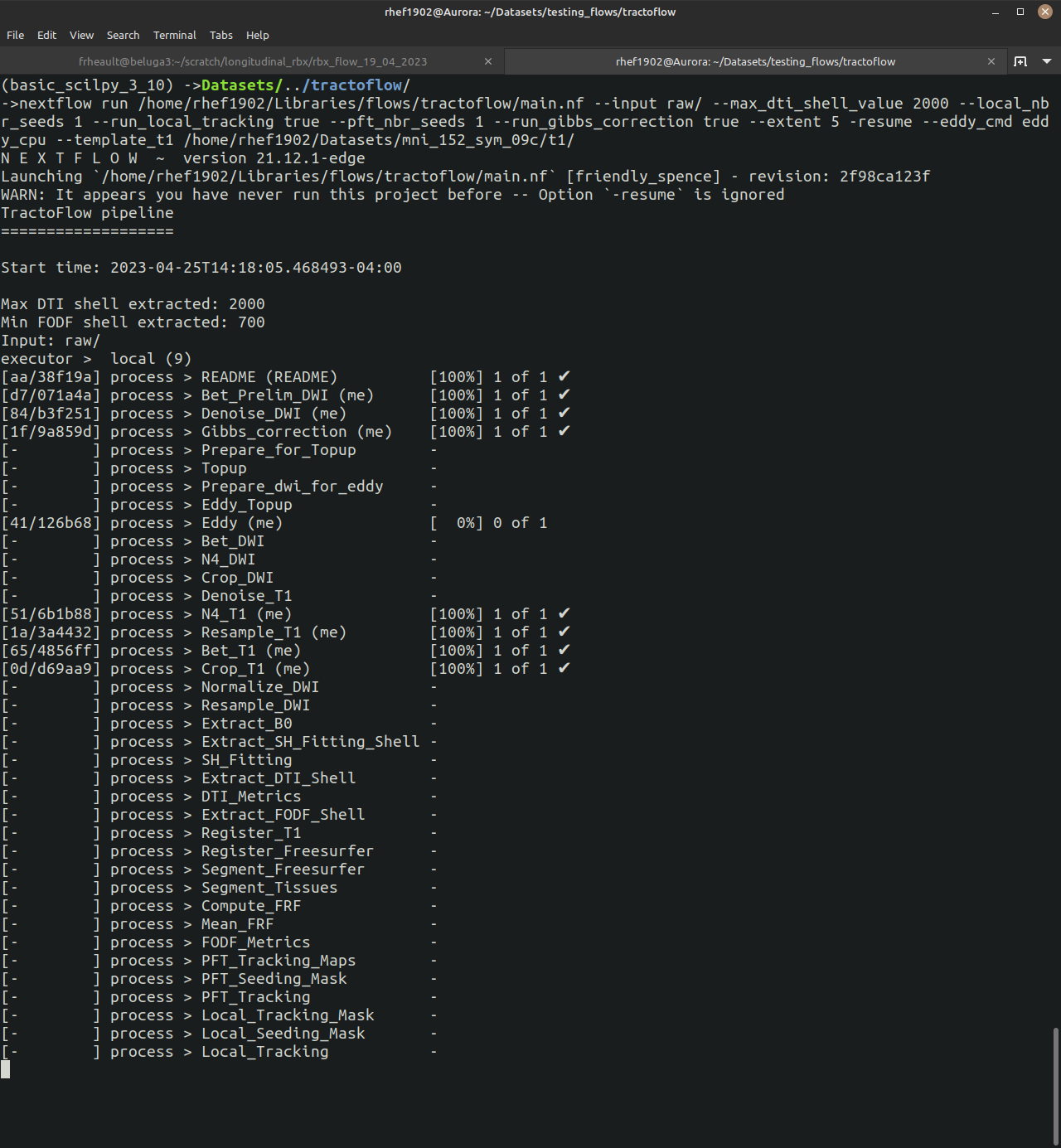
Once you get the pipeline running, you can follow along easily. You will see all the processes that have to be run as well as how many subjects are waiting/completed.
- When completed you will see a few directories created by the pipeline:
results/contains symbolic links (towork/) of all output data organized with the name of the process they came from. All files start with SID__ (subject id) as a nomenclature.work/contains all of the processing data and scripts, but organized with hash that allow -resume to work.nextflow/contains a bunch of cached file needed by Nextflow*.html(if using-with-report)
If your pipeline crashs, you should see something like:
[b7/94b24c] NOTE: Process `Bet_Prelim_DWI (me)` terminated with an error exit status (2) – Execution is retried (1)
This means the process Bet_Prelim_DWI for the subject me crashed, and execution was retried once (1). To see the error message you should use cat work/b7/94b24c at this point you press tab for auto-completion (this is unique to every run, subject, process). Then your full command will look like cat work/b7/94b24c5dc1c29e689be628086f3b0/.command.log, and it will display the error encountered.
Understanding Outputs
Once completed, all pipelines will produce a results/ folder with all subjects, and within each all processes will be in there. In our example, Tractoflow had a single subject named me. The folders are in alphabetical order, not in order of completion.
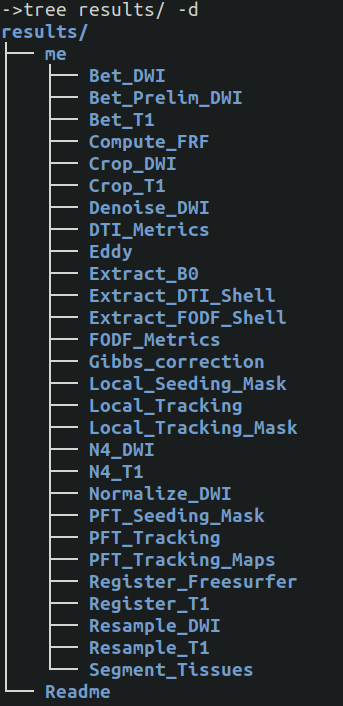
When run successfully, the organization is very intuitive. All files have a standard naming convention and should always exists.
Finally, no matter which Nextflow pipeline you are using. The results/ directly will always be symbolic links, so make sure to never delete the work/ folder before copying it safely (i.e. cp -rL results/ results_no_links).
Using it with Containers
If you’re handling big datasets in scientific research, you know how quickly things can get overwhelming. One great solution is to use Nextflow with containers – not only does this simplify your workflows by keeping everything in one place, but it also provides an extra layer of reproducible and scalable computing infrastructure.
If you’re serious about efficient, reproducible scientific research, then Nextflow with Containers is definitely worth your time.
With Nextflow you can use the flag -with-docker scilus/docker-tractoflow:2.1.1 (hypothetical version) to use a specific container on your computer or in this case, fetch a docker from our online DockerHub. You can also use -with-singularity /PATH/TO/DATA/scilus_tractoflow_2.1.1 to use a local singularity, this works best on ComputeCanada since we do not have sudo privilege.
In both case, the goal is to keep an exact trace of which code of which version you used. Knowing your version of the Nextflow Pipeline and the Containers used will help you document what you launched without writing the version of hundreds of tools and libraries. It will make it a lot easier to re-launch everything (if needed) a few months later.