RecobundlesX
Recobundles is a tool to separate your wholebrain tracking result into different bundles divided into separate files. Dipy has a published version, called Recobundles. It is single atlas and single parameter while our version (which we call RecobundlesX) is multi-atlas and multi-parameter, and shown to be more robust in Rheault 2020 (PhD thesis, chapter 4, https://savoirs.usherbrooke.ca/handle/11143/17255). An atlas is available on zenodo (models, config and reference). There is a Nextflow pipeline for this tool, available on Github
You can find the description of Dipy single atlas / single parameter here. We also provide a convenient wrapper for that version in scil_recognize_single_bundle.py which is simpler to use. For multiple bundles however, we highly recommend scil_recognize_multi_bundle.py (i.e. RecobundlesX).
Here is an example of how to run RecobundlesX. In the following script, we take the example of data that has been preprocessed using Tractoflow. tractoflow_folder is your root, which should contain tractoflow’s results for each subject. This script will create a RecobundlesX folder inside each subject’s directory.
Before running, read carefully the script’s documentation (-h) in Scilpy.
You will need ANTs
tractoflow_folder=my_dir
subject_list=my_subject_list.txt
models_path=YOUR_PATH # Ex: hcp_models/
model_T1=THE_T1 # Ex: ${models_path}/mni_masked.nii.gz
model_config=JSON_FILE # Ex: ${models_path}/config_python.json
# Filtering options (in mm). Change as needed.
minL=20
maxL=200
# RecobundlesX options. Change as needed.
nb_total_executions=9 # len(model_clustering_thr) * len(bundle_pruning_thr) * len(tractogram_clustering_thr) = max total executions (see json).
thresh_dist="10 12" # Whole brain clustering threshold (in mm) for QuickBundles.
processes=6 # Number of thread used for computation.
seed=0 # Random number generator initialisation.
minimal_vote=0.5 # Saving streamlines if recognized often enough.
while IFS= read -r subj; do
echo "Running subject ${subj}"
# Defining subj folders
subj_folder=${tractoflow_folder}/${subj}
# Defining inputs
subj_trk=${subj_folder}/Tracking/${subj}__local_tracking*.trk
subj_T1=${subj_folder}/Register_T1/${subj}__t1_warped.nii.gz
###
# Registering model on subject (using ANTS)
# -d=image dimension,
# -f=fixed image, m=moving image
# -t: transformation a = rigid+affine
# -n = nb of threads
# This should create 3 files : model_to_subj_anat0GenericAffine.mat,
# model_to_subj_anatInverseWarped.nii.gz and model_to_subj_anatWarped.nii.gz
###
model_to_subj=${rbx_folder}/model_to_subj_anat
antsRegistrationSyNQuick.sh -d 3 -f ${subj_T1} -m ${model_T1} -t a -n 4 -o ${model_to_subj}
###
# Cleaning tracking file to make sure it is not too big.
# Recobundles is already long enough :) .
###
subj_filtered_trk=${subj_folder}/Tracking/${subj}__tracking_filteredLength.trk
scil_filter_streamlines_by_length.py --minL $minL --maxL ${maxL} ${subj_trk} ${subj}_filtered_trk
###
# Scil's Recobundle
# processes = nb of threads.
# Seed = rnd generator seed.
# inverse is to use the inverse affine
###
mkdir ${rbx_folder}/multi_bundles
scil_recognize_multi_bundles.py ${subj}_filtered_trk ${model_config} ${atlas_dir} ${affine} \
--out_dir ${rbx_folder}/multi_bundles \
--processes ${processes} --seeds ${seed} \
--minimal_vote_ratio ${minimal_vote} \
--log_level DEBUG --inverse -f
done < ${subject_list}
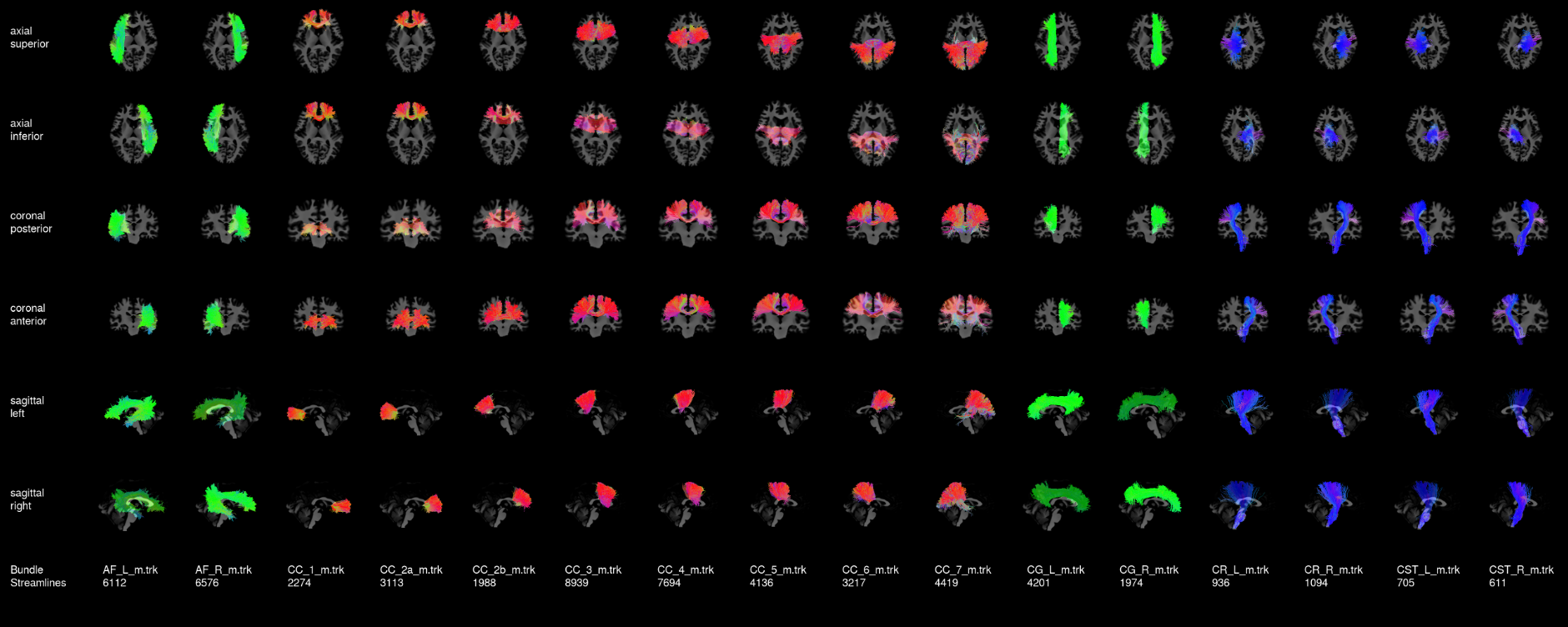
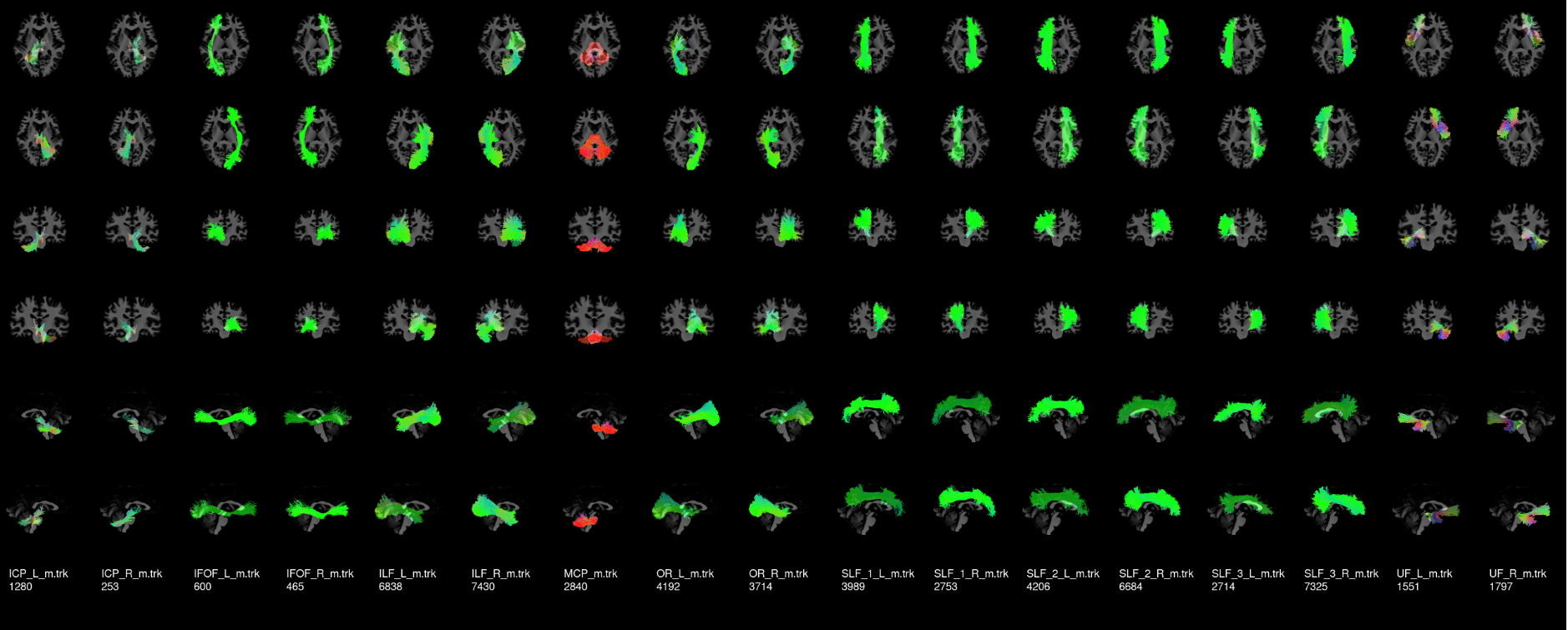
To visualize your results for one subject, here is a nice tool:
anat=YOUR_ANAT rbx_folder=YOUR_RBX_FOLDER scil_visualize_bundles_mosaic.py ${anat} ${rbx_folder}/*.trk mosaic.png
Here is a nice example to help your compare your results. This was created from a HCP subject.